
DOP50 The landscape of somatic mutations in non-neoplastic IBD-affected colon
S. Olafsson1, R.E. McIntyre1, T. Coorens2, T. Butler2, P. Robinson2, H. Lee-Six2, M. Sanders2, K. Arestang3, C. Dawson3, M. Tripathi4, K. Strongili3, Y. Hooks2, M.R. Stratton2, M. Parkes3, I. Martincorena2, T. Raine3, P.J. Campbell2, C.A. Anderson1
1Department of Human Genetics, Wellcome Trust Sanger Institute, Hinxton, UK, 2Cancer, Aging and Somatic Mutations, Wellcome Trust Sanger Institute, Hinxton, UK, 3Department of Gastroenterology, Addenbrooke’s Hospital, Cambridge, UK, 4Department of Histopathology, Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK
Background
Under normal physiological conditions, colonic crypts accrue ~40 somatic mutations for every year of life. That somatic mutations contribute to the development of cancer is well established, but their patterns, burden and functional consequences in diseases other than cancer have not been extensively studied and our understanding of the effects of chronic inflammation on the mutational profile and clonal structure of the colon is limited. Here, we investigated how the recurrent cycles of inflammation, ulceration and regeneration seen in IBD impact the mutational and clonal structure of intestinal epithelia.
Methods
We isolated and whole-genome sequenced ~400 individual colonic crypts from 46 IBD patients and compared these to ~400 crypts from 41 non-IBD controls. We compared the mutation burden, mutational signature exposure, clonal structure and cancer driver mutation landscape in crypts from actively and previously inflamed regions with crypts dissected from controls.
Results
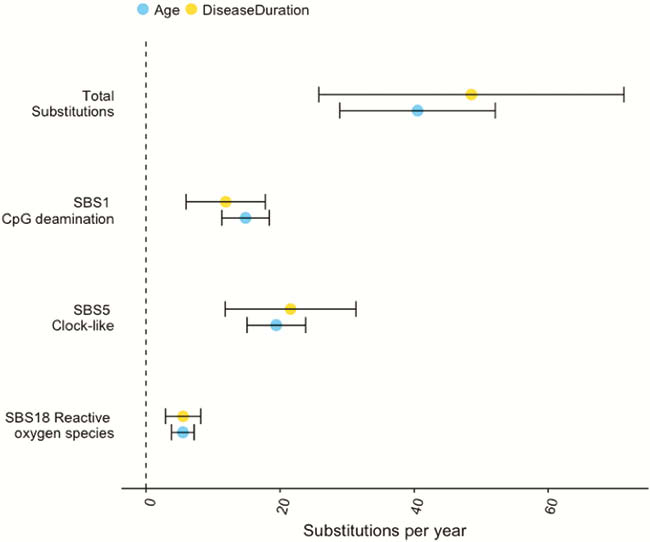
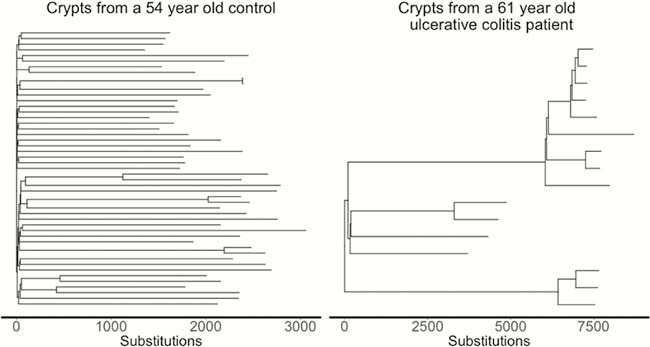
We estimated the base substitution rate of affected colonic epithelial cells to be doubled after IBD onset. This change was primarily driven by acceleration of mutational processes ubiquitously observed in normal colon (Figure 1), and we did not detect an IBD-specific mutational process. In contrast to the normal colon, where clonal expansions outside the confines of the crypt are rare, we observed widespread millimetre-scale clonal expansions, even in the absence of mutations in
Conclusion
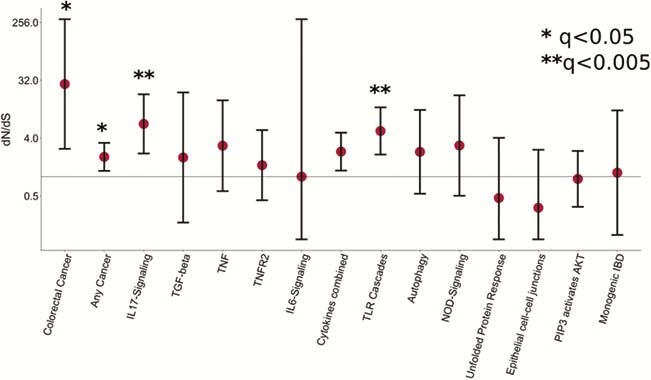
Our results provide new insights into the consequences of chronic intestinal inflammation on the mutational profile and clonal structure of colonic epithelia. We identify the mutagens driving the increase in mutation burden and mutations which are under positive selection in the context of inflammation. Our results suggest that studying somatic mutations in the colon can reveal putative drug targets and pathogenic mechanisms for IBD.