
DOP79 Dose escalation of upadacitinib in Crohn’s disease patients with an inadequate response: Data from the randomised CELEST study
W. Sandborn MD1, J. Panés2, L. Peyrin-Biroulet3, E. Louis4, S. Greenbloom5, W. Zhou6, Q. Zhou7, J. Liu8, A.P. Lacerda6
1Division of Gastroenterology, University of California San Diego, La Jolla, USA, 2Inflammatory Bowel Diseases Unit, Hospital Clínic Barcelona- IDIBAPS, CIBERehd, Barcelona, Spain, 3Department of Gastroenterology, University of Lorraine, Nancy, France, 4Department of Gastroenterology, University Hospital CHU of Liège, Liège, Belgium, 5Department of Gastroenterology, Toronto Digestive Disease Associates, Toronto, Canada, 6Global Pharmaceutical Research and Development, AbbVie Inc., North Chicago, USA, 7Data and Statistical Sciences, AbbVie Inc., North Chicago, USA, 8Pharmacovigilance and Patient Safety, AbbVie Inc., North Chicago, USA
Background
Patients with Crohn’s disease (CD) may lose response to treatment, requiring dose adjustments. This analysis from the CELEST study evaluated efficacy and safety in patients with CD who required an escalated dose of upadacitinib (UPA), an oral selective JAK1 inhibitor.
Methods
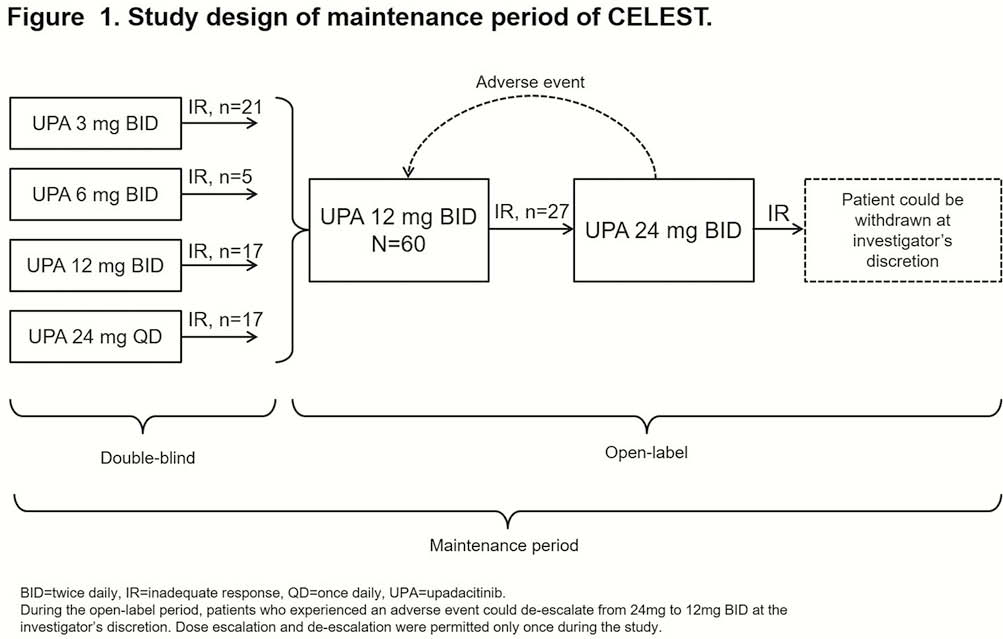
CELEST (NCT02365649) was a placebo-controlled phase 2 study of adults with moderate-to-severe CD refractory to immunosuppressants/biologics. Patients were randomised to placebo or UPA 3, 6, 12, or 24 mg twice daily (BID) or 24mg once daily (QD) for 16 weeks (weeks), followed by a 36-week double-blind maintenance period to receive 3, 6 or 12mg BID or 24mg QD (Figure 1). Patients who met criteria for inadequate response (average daily liquid/soft stool frequency [SF] >2.2 or average daily abdominal pain score >1.8, and increase in hs-CRP >1 mg/l from baseline [BL] or hs-CRP ≥5 mg/l at previous or current visit) at or after week 20 were eligible to receive open-label (OL) therapy with UPA 12mg BID. Patients with inadequate response at or after 4 weeks of OL 12 mg BID could further escalate to OL 24 mg BID. Data were analysed using descriptive statistics of clinical/endoscopic improvement, change from BL in hs-CRP and faecal calprotectin (FC), and AEs at week 52.
Results
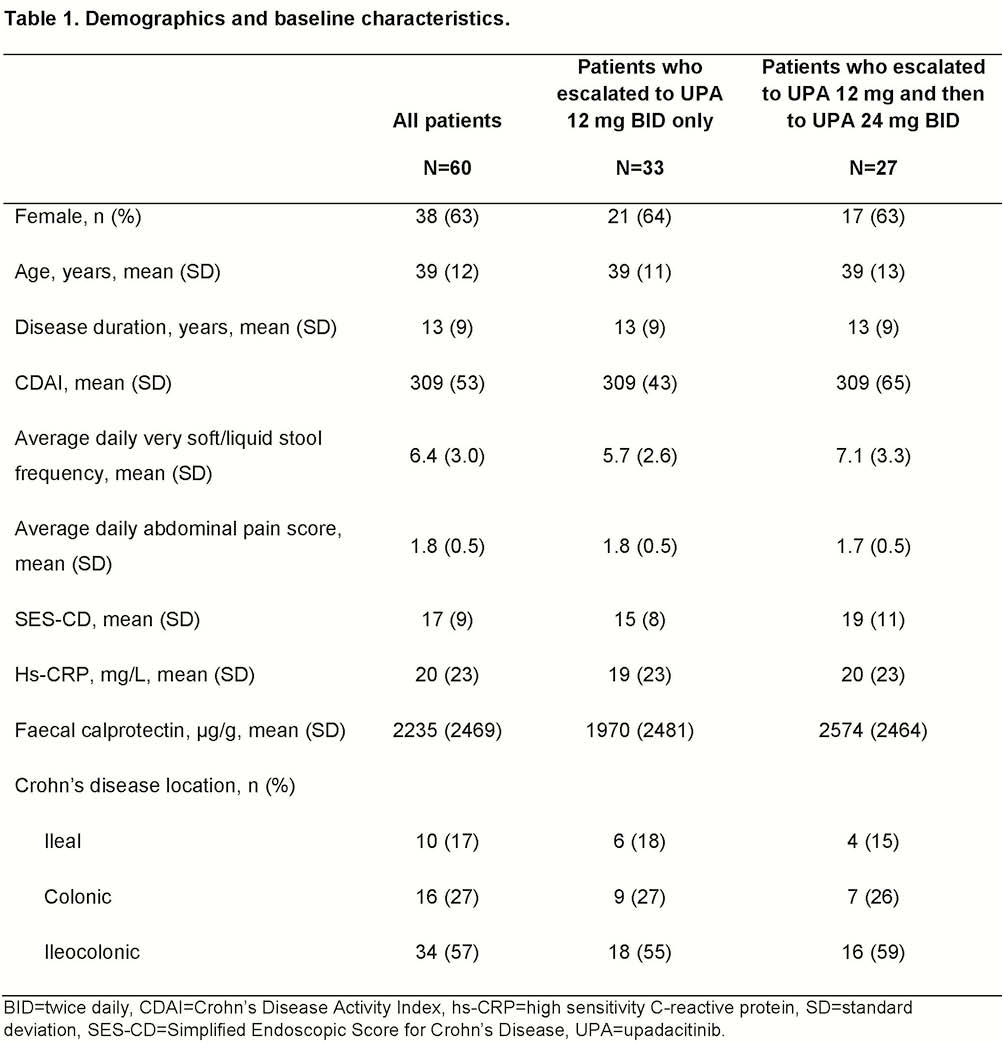
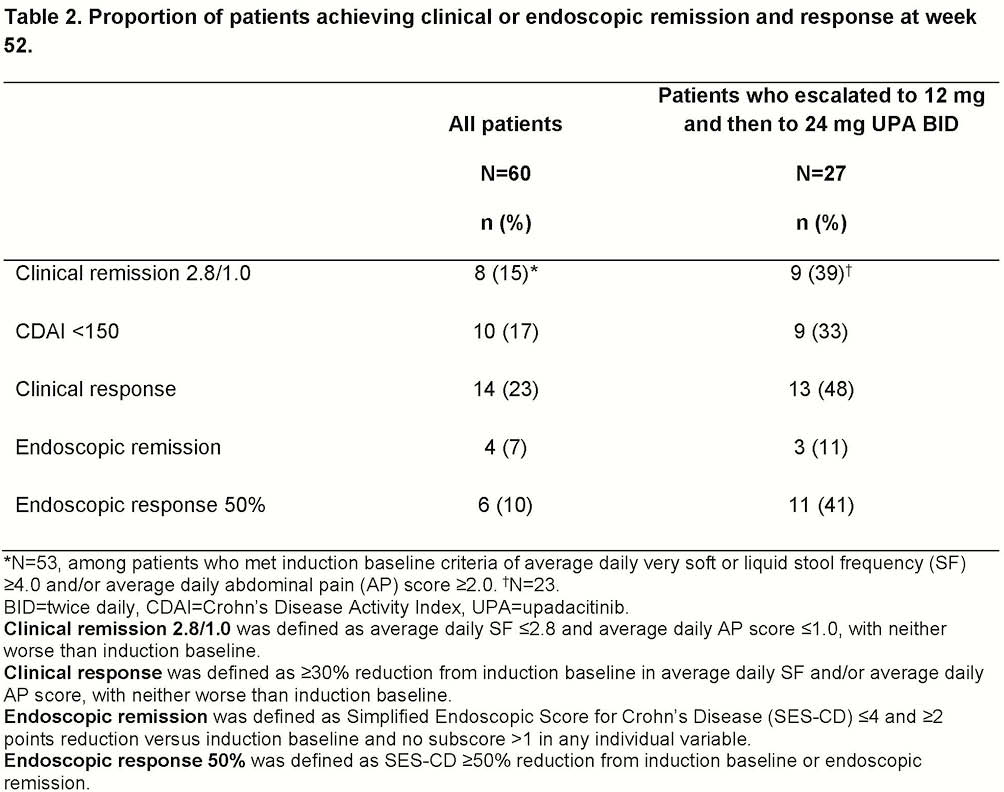
Among 220 randomised patients, 180 were re-randomised into the maintenance period; 60 had inadequate response and received OL 12 mg BID. Of the 60 patients, 25 (42%) were in clinical response at week 16. After receiving OL 12 mg, 27 patients (45%) required escalation to OL 24 mg BID. A higher proportion of patients who received OL 24 mg received 2 or more prior TNF antagonists, and had higher BL FC, SF, and Simplified Endoscopic Score for CD (SES-CD) compared with patients who remained on OL 12 mg BID (Table 1). At week 52, 15% of patients who received OL 12 mg achieved clinical remission and 10% achieved endoscopic response. Of the patients who escalated to OL 24 mg, 39% achieved clinical remission and 41% were in endoscopic response at week 52 (Table 2). At week 52, change from BL in FC and hs-CRP was −2123 ± 2475 µg/g and −5.1 mg/l, respectively, for patients who received OL 24 mg vs. −423 ± 2953 and −6.4 mg/l for OL 12 mg. Serious infection was reported in 3 patients (2 receiving OL 12 mg and 1 receiving OL 24 mg). Two nonserious herpes zoster events were reported (1 on each dose), but neither event led to discontinuation of UPA. No malignancies, cardiovascular events, thromboembolic events, intestinal perforations, tuberculosis, or deaths were reported for patients who dose-escalated.
Conclusion
Patients with non-response or loss of response during CELEST gained clinical remission and endoscopic response with UPA 12 mg BID or further escalation to 24 mg BID. No new safety risks were observed for either dose of UPA, consistent with double-blind CELEST results.