
OP11 Exposure to an inflammatory mix re-induces inflammation in organoids of ulcerative colitis patients, independent of the inflammatory state of the tissue of origin
K. Arnauts1,2, B. Verstockt1,3, J. Sabino1,3, S. Vermeire1,3, C. Verfaillie2, M. Ferrante1,3
1Department of Chronic Diseases, Metabolism and Ageing, KU Leuven, TARGID, Leuven, Belgium, 2Department of Development and Regeneration, Stem Cell Institute Leuven, KU Leuven, Leuven, Belgium, 3Department of Gastroenterology and Hepatology, KU Leuven, University Hospitals - Leuven, Leuven, Belgium
Background
Patient-derived intestinal organoids provide a powerful tool to unravel mechanisms underlying inflammatory bowel disease (IBD). Recently, we showed that organoids derived from inflamed regions in ulcerative colitis (UC) patients lose their inflammatory phenotype during
Methods
Biopsies were obtained from 8 patients with active UC (endoscopic Mayo score of ≥2), both in inflamed and non-inflamed regions, and in 8 non-IBD controls. Crypts were isolated and cultured as organoids for at least four weeks. Organoids were subjected to a predefined inflammatory mix (MIX: 100 ng/ml TNF-α, 20 ng/ml IL-1β, 1 µg/ml Flagellin) or medium only (CTRL) for 24 h (Figure 1). RNA was extracted from organoids for RNA sequencing by Lexogen QuantSeq for Illumina. Differential gene expression and pathways were studied through DESeq2 and ingenuity pathway analysis (false discovery rate <0.05).
Results
Prior to inflammatory stimulation, principal component analysis (PCA) demonstrated separate clustering between organoids derived from non-IBD controls and UC patients. Exposure to the inflammatory mix induced transcriptional activation of inflammatory genes (
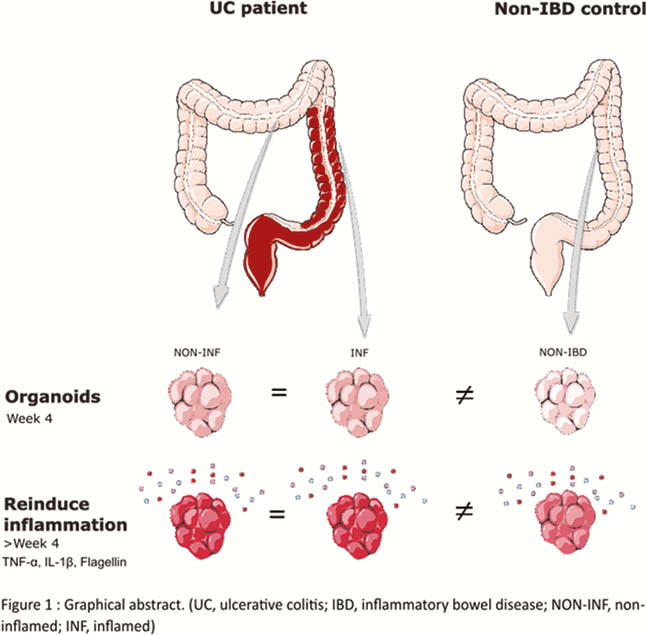
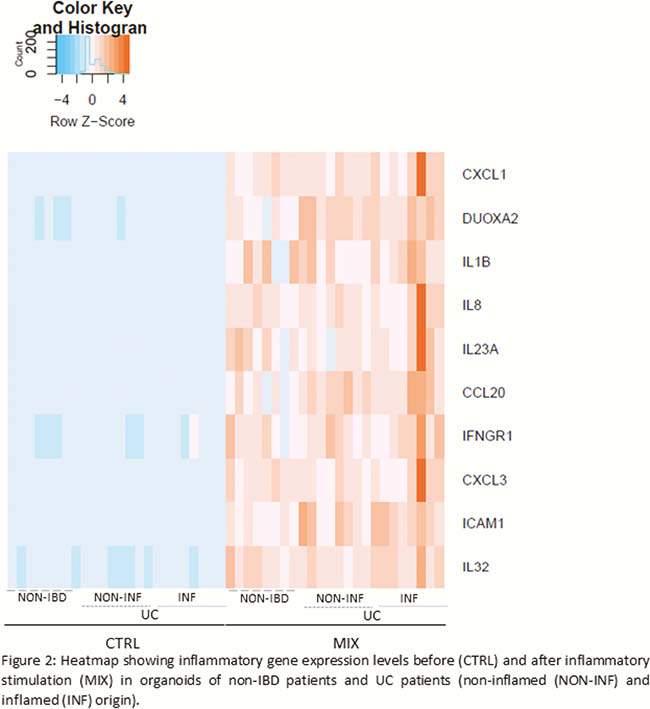
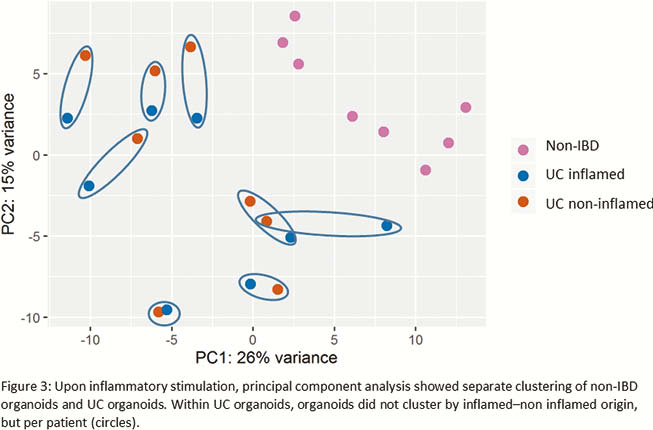
Conclusion
Inflammation can efficiently be (re-)induced in organoids from both UC and non-IBD origin. However, a different response was observed between organoids of non-IBD and UC origin. Of note, in UC organoids, the state of inflammation in the source tissue was irrelevant. In conclusion, we showed that it is essential to re-induce inflammation in patient-specific organoids, but there is no need to obtain biopsies from inflamed regions.
Arnauts K. (2019).