I. Hageman1, A. Li Yim2, V. Joustra3, M. Ghiboub1, K. Gecse3, A. Te Velde1, G. D’Haens3, C. Paulusma1, P. Henneman2, W. De Jonge1
1Academic Medical Center AMC University of Amsterdam, Tytgat Institute for Liver and Intestinal Research, Amsterdam, The Netherlands, 2Department of Clinical Genetics, Academic Medical Center AMC University of Amsterdam, Amsterdam, The Netherlands, 3Department of Gastroenterology and Hepatology, Academic Medical Center AMC University of Amsterdam, Amsterdam, The Netherlands
Background
SP140 (Speckled 140 KDa) encodes an epigenetic reader protein with an immune restricted expression, that binds to epigenetically modified (acetylated and methylated) histones and thereby regulates expression of large gene sets, including pro-inflammatory cytokines, in innate immune cells. SP140 is implicated in CD because single nucleotide polymorphisms, as well as defective protein function are associated with CD and marks anti-TNF response. Through a genome-wide methylation screen of Crohn’s disease (CD) patients peripheral blood, we identified two hypermethylated positions in SP140 locus associated with CD patients. We hypothesise that this DNA hypermethylation at the SP140 locus controls SP140 expression in CD patients contributing to their colitis development.
Methods
To address the role of SP140 DNA methylation, we used CRISPR ‘dead’ Cas9 (dCas9) epigenome-editing for specifically adding methyl groups (dCas9-DNMT) or removing methyl groups (dCas9-TET) in monocyte cell line THP1. We developed guide RNAs complementary to the gene expression regulatory region of the SP140 gene. With lentiviral delivery, we transduced THP-1 cells with guide RNA-lentiviruses, and with dCas9-DNMT or dCas9-TET lentiviruses. We assessed the level of SP140 methylation using bisulphite Sanger sequencing and the effect of methylation intervention of SP140 using qPCR and ELISA for SP140, IL-6, TNFα, IL-1β.
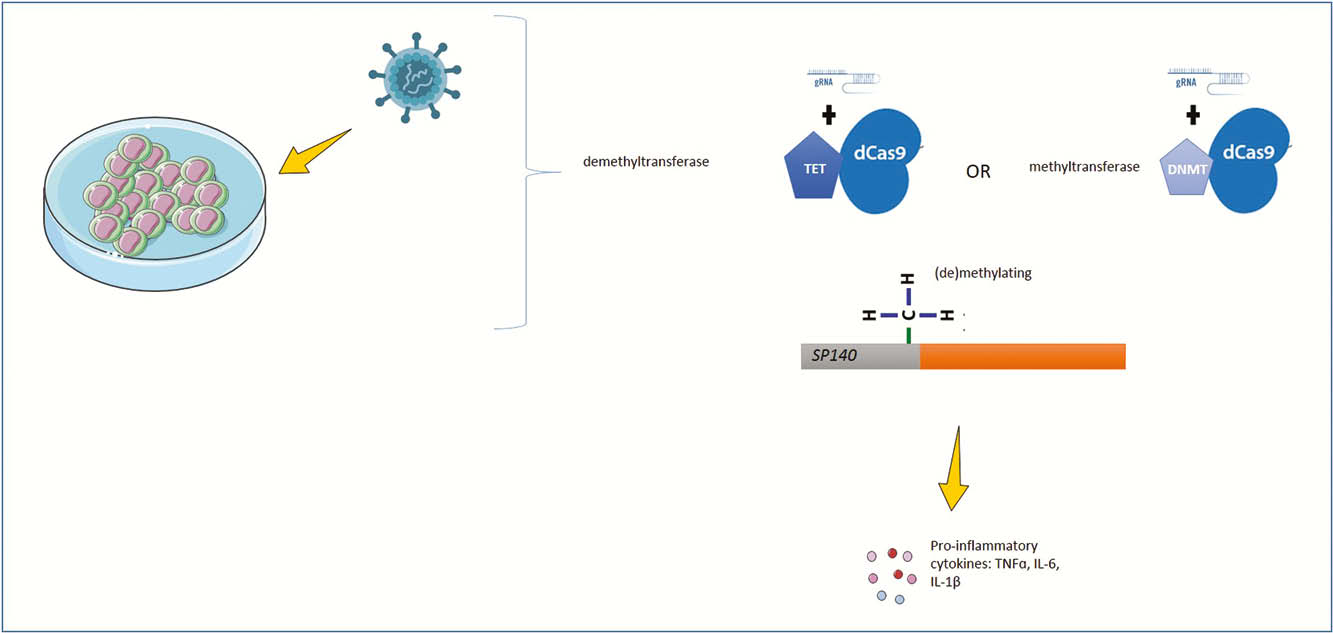
Results
We observed that SP140 gene in THP-1 cells under control conditions contained little methylated CpG sites. We induced sp140 hypermethylation through transduction of dCas9-DNMT. We validated hypermethylation of the two SP140 CpGs in transduced THP1 cells, thus mimicking the observed hypermethylation in CD patients cells. SP140 hypermethylation in THP1 cells polarised into M1 macrophages and stimulated with lipoteichoic acid (TLR-2 ligand), displayed a decrease of TNFα and (p = 0.042) protein levels. Similarly, we showed a decrease of TNFα (p = 0.02) and IL-6 (p = 0.03) protein release after transduction dCas9-DNMT and stimulation with LPS or Zymosan (TLR-2/4 ligand).
Conclusion
In this study, we demonstrated that editing SP140 gene methylation through CRISPR-dCAS9 technology allows modelling of the relevance of epigenetic marks for CD aetiology. Through methylome editing, we could affect the expression of CD-associated pro-inflammatory genes. Our dCas9 technique will allow us to investigate the role of DNA-methylation in the aetiology of CD.
