
P702 Faecal Microbiota Transplantation Sculpts the faecal and mucosal microbial and metabolomic profiles in patients with Ulcerative Colitis
Bajaj, A.(1);Markandey, M.(1);Singh, M.(1);Virmani, S.(1);Vuyyuru, S.K.(1);Kante, B.(1);Thirunavukkarasu, R.(2);Verma, M.(1);Kumar, P.(1);Kshetrapal, P.(2);Makharia, G.(1);Gupta, D.(3);Kedia, S.(1);Ahuja, V.(1);
(1)All India Institute of Medical Sciences, Department of Gastroenterology and Human Nutrition, New Delhi, India;(2)Translational Health Science and Technology Institute, NCR Biotech Science Cluster, Faridabad 121001, India;(3)International Centre for Genetic Engineering and Biotechnology ICGEB, International Centre for Genetic Engineering and Biotechnology ICGEB, New Delhi, India;
Background
Gut microbiota modulation via faecal microbiota transplantation (FMT)is known to induce long lasting clinical as well as endoscopic remission in patients with UC. The present study aims to identify microbial and metabolomic changes in the faecal as well mucosal niches, in response to FMT in patients with UC.
Methods
28 patients with mild-moderate UC and 16 non-IBD controls were enrolled. Patients were given a weekly infusion of pooled-multidonor-FMT, for 8 weeks, while maintaining a uniform dietary pattern. We collected paired stool and recto-sigmoidal biopsysamples from patients with UC pre-FMT (n=28), post-FMT (n=10) and controls (n=16). 16S (V3-V4) rRNA sequencing and LC-MS based untargeted metabolomics was performed. α and β diversity analysis and differential abundance (DA) analysis of microbiota was performed using DeSeq2 R package. Differential metabolite peaks between the three groups were identified via volcano plot and annotated using LipidMaps, HMDB, Kegg and Metlin databases. (Fig.1)
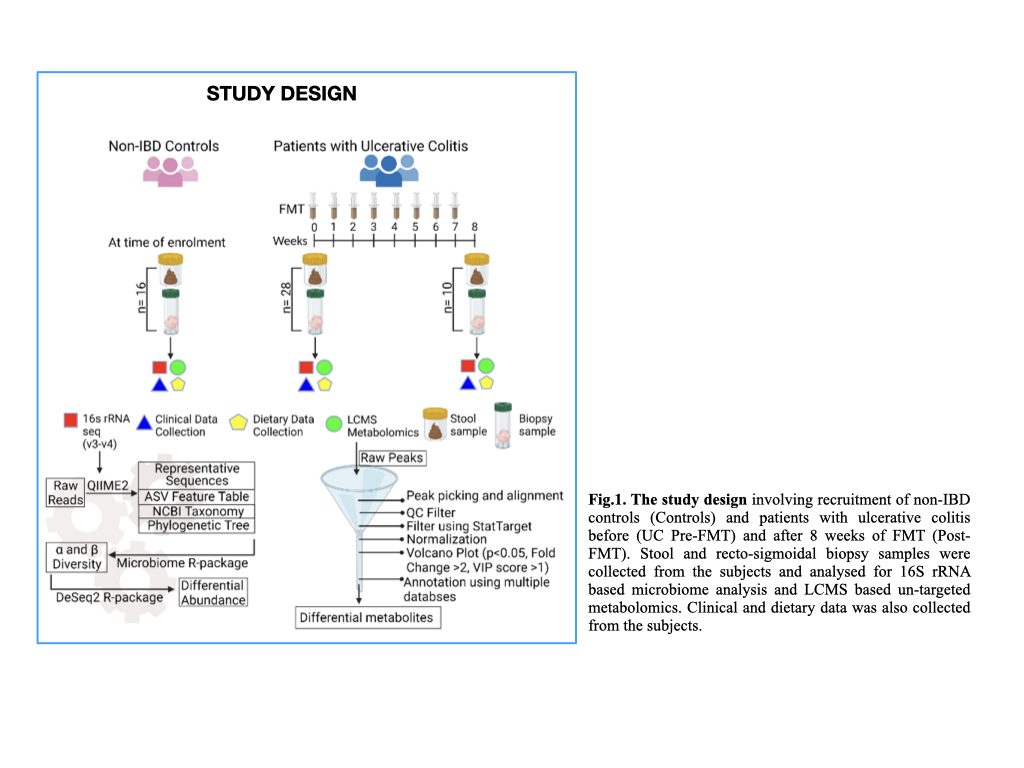
Results
α-diversity of patients with UC was not significantly different from that of controls, in both sample matrices. Post-FMT α-diversity of faecal samples differed significantly from the Pre-FMT group, however in mucosal tissue, the difference was non-significant. β-diversity indices were significant in UC pre-FMT vs post-FMT populations for both matrices (Fig.2A and B). DA analysis identified faecal and mucosal controls and UC post-FMT samples to be significantly similar [ANOSIM R=0.07; p=0.1 (faecal) and R=0.02; p=0.315 (mucosal)]. Analysis of core microbiota revealed diminished faecal microbial core in UC, with no major UC-associated loss of mucosal core members, showing relatively conserved nature of mucosal microbiota. FMT restored microbial core along both niches.
UC pre-FMT samples displayed increased abundance of Firmicutes (Limosilactobacillus, Lactobacillus, Streptococcus, Veilonella and Sphingomonas) and Proteobacteria (Methylobacterium) in faecal samples, while mucosa-associated microbiota displayed increased abundances of Firmicutes (Streptococcus, Limosilactobacillus, Ligilactobacillus, Eubacterium, Enterococcus, etc), Bacteroidetes (Parabacteroides and Alistipes) and Proteobacteria (Burkholderia)(Fig.3). Distinct functional classes of metabolites, including bacterial cell wall and QS molecules, membrane lipids, metabolism of vitamins, long and medium chain fatty acids, bile acids, and tryptophan etc, were also found to be altered between the three groups in both sample matrices(Fig.4)
UC pre-FMT samples displayed increased abundance of Firmicutes (Limosilactobacillus, Lactobacillus, Streptococcus, Veilonella and Sphingomonas) and Proteobacteria (Methylobacterium) in faecal samples, while mucosa-associated microbiota displayed increased abundances of Firmicutes (Streptococcus, Limosilactobacillus, Ligilactobacillus, Eubacterium, Enterococcus, etc), Bacteroidetes (Parabacteroides and Alistipes) and Proteobacteria (Burkholderia)(Fig.3). Distinct functional classes of metabolites, including bacterial cell wall and QS molecules, membrane lipids, metabolism of vitamins, long and medium chain fatty acids, bile acids, and tryptophan etc, were also found to be altered between the three groups in both sample matrices(Fig.4)
Conclusion
FMT efficiently restores beneficial bacterial populations and related metabolic pathways in faecal as well the relatively reserved mucosal microbial niches, which may pave way for induction of remission in patients with UC.


